Using mouse models of cancer to identify cancer cell vulnerabilities and target them for therapy
Deciphering the gut microbiome regulation of p53 mutations in intestinal cancer development
Our lab has recently discovered a surprising tumor suppressive effects of the notorious mutant p53 (Kadosh et al., Nature 2020) and highlighted a remarkable functional plasticity of mutant p53, as well as the instructive role played by the microbiota in molding this plasticity. We saw a dichotomous effect of hotspot mutant p53 (mutp53) in two Wnt-driven colorectal cancer mouse models: counteracting tumorigenesis in the proximal gut while enhancing tumorigenesis in the distal gut. We proved that mutant p53 activity in gut epithelia is influenced by local production of microbial metabolites; the switch of p53 from tumor suppressor to oncogene is location-dependent and is impacted by microbiome-derived gallic acid. Our unexpected findings challenge the common view of autonomous effects of cancer driving mutations and underscore their critical dependence of certain mutations on the gut microbiome.
This is in line with the evidence for a very low frequency of cancer at the small intestine in humans, 50 times less than cancer in the colon, considering that the small intestine is 3 times longer than the colon. Notably, somatic p53 mutations are fairly prevalent in some normal human organs and seem to promote clonal selection and expansion in healthy human skin and in the aging esophagus. It is therefore possible that wheres germline TP53 mutations are detrimental and predispose to early onset of different cancer types, localized somatic p53 mutations, especially at an older age, may be beneficial, protecting externally-exposed organs from damaging agents. We have started looking for evidence supporting this hypothesis in mouse models.
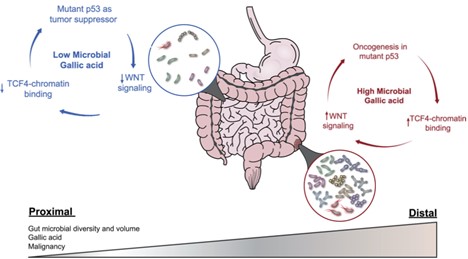
Adapted from: White, M. G., & Wargo, J. A. (2020). Gut Microbes' Impact on Oncogenic Drivers: Location Matters. Molecular Cell, 79(6), 878-880.
Another aim of our future work in this project is to reveal the interrelationship of the microbiome with the variety of TP53 mutants in the process of CRC development. Using the TP53 mutant library constructed by Moshe Oren we will screen adenoma organoids derived from APC-mutated mice, to identify which TP53 mutations exhibit tumor suppressor activity. Bioinformatic and structure-based analysis may reveal rules classifying mutp53 molecules having tumor suppressor properties, and the final validation of these properties will take place in mouse models of intestinal cancer. Using first the intestinal organoid system with defined genotypes and then mouse models, we will identify microbiota species and microbiota-derived factors that convert a mutp53 with tumor suppressor properties to an oncogene. Our collaborator Naama Geva-Zatorsky has a large collection of bacterial culture media and metabolites which will be used for this purpose. We will identify microbiota products that modify the structure-function of mutp53, the epigenome of mutant-bearing cells, or host cell signaling affecting mutp53 function and then determine the effects of pro-tumorigenic bacterial products on mutp53-mediated tumorigenesis in intestinal tumor mouse models. Finally, with our colleagues at the Hadassah Medical Center we will identify in patients bearing high risk for CRC (such as inflammatory bowel disease patients) specific gut bacteria and/or gut microbiota composition that may promote the oncogenic switch of mutp53 in premalignant gut tumors. We will interface this data with dietary information towards cancer risk assessment and prevention.
Studying the mechanisms that promotes primary epithelial invasion to initiate cancer
The transition of tumor cells from the non-invasive phenotype to invasive phenotype of converts a benign tumor that can be successfully treated surgically to a malignant form of cancer. Metastatic cancer is the leading cause of cancer-related death and the primary step for cancer metastasis is cell invasion, yet it is not clear what are the initial key steps that promote this process. As cancer is a slow process that is not readily amenable to direct microscopic observation, the mechanism of invasion and cellular dynamics therein are less studied. We have shown before that upon deletion of both CKIα and p53 in the mouse gut (CKIαΔgut/p53Δgut), intestinal homeostasis is disrupted and mice develop widespread high-grade dysplasia and intramucosal carcinomas (Elyada et al, 2011). Moreover, we found that compared with CKIαΔgut mice, in CKIαΔgut/p53Δgut mice there was an upregulation of a set of genes denoted PSIS genes (p53-suppressed invasiveness signature), that are associated with diverse invasiveness functions. One of these PSIS genes is Prox1, which serves as a marker for invasion in the gut. Interestingly, we also showed that treatment of CKIαΔgut/p53Δgut mice with non-steroidal anti-inflammatory drugs (NSAIDs), led to the suppression of PSIS genes and the invasive mouse phenotype. This indicated that inflammatory mediators may synergize with p53 loss in upregulating PSIS genes and invasiveness induction. One of these pro-inflammatory mediators, Ptges1 which its product, PGE2, is associated with invasiveness in many types of cancer, was upregulated in CKIαΔgut mice, but reduced in CKIαΔgut/p53Δgut mice. Using Prox1-GFP reporter mice, we intend to characterize early mechanisms that promote the invasion process in the gut and understand whether and how Ptges1 is involved in this process. Prox1 reporter mice have been generated for this purpose and will be used to analyse Prox1+ early invasive gut epithelial cells and determine their unique properties compared to Prox1-negative cells. This study may also answer important questions about evolution of tumor heterogeneity, clonality and effect of the microenvironment.
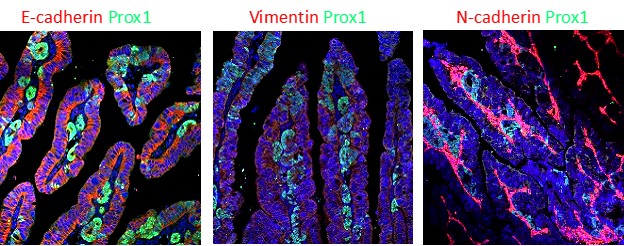
Seeds of cancer? Transformed epithelial cells (Prox1+ cells stained in green) are taking the first steps of invasion and metastasis in the epithelium of small intestine.
Developing small molecule multi-kinase inhibitors as novel anti-leukemia agents and beyond
Acute myeloid leukemia (AML) is a relatively infrequent cancer, yet accounts for 2 percent of all cancer death. Despite some recent remarkable developments in treating certain AML types, the standard therapy of the disease hasn't changed much in the past 50 years. AML commonly harbors non-mutated p53, but the tumor suppressor protein is frequently inactivated due to enhanced activity of its negative regulators, particularly Mdm2. These regulators complement aberrant epigenetic reprograming in protecting the malignant cells; their elimination may enhance cancer vulnerability by increasing the sensitivity to chemotherapy.
A breakthrough in our laboratory was the discovery that CKIα ablation is also an effective way of activating p53 (Elyada et al., Nature, 2011). As a result, we embarked on the development of small molecule inhibitors of CKIα and produced the first prototype high affinity inhibitors of this CKI isoform. Testing these inhibitors in animal models of AML showed profound therapeutic responses in both genetically modified mouse models and patient derived xenografts. Consequently, the lead compound A51 we developed has recently been approved as an investigational new drug (IND) by the FDA and will soon be tested in phase I clinical trial in treatment-refractory and relapsing MDS and AML patients (
Link).
As a mechanism of action, we found that CKIα inhibitors having the best anti-leukemic activity also target the transcriptional kinases CDK7 and CDK9, which phosphorylate RNA Pol II CTD at Ser5 and Ser2. Inhibition of these kinases strongly affected specific gene-regulatory clusters known as super-enhancers (SEs), having a major role in driving oncogene expression. One common form of cancer cell vulnerability is transcriptional addiction, often associated with a robust transcriptional thrust mediated by SEs. SEs have recently been highlighted as attractive targets for epigenetic therapy of cancer, particularly in hematological malignancies, although no such therapy has yet demonstrated an outstanding clinical benefit, due to toxicity and low efficacy. Considering these unique properties, we chose to term this kind of inhibitors oncodestroyers (ODs), standing for single molecules which by co-targeting several critical oncogenic hubs maximize cancer cell death.
To further elucidate the mechanism of action of this kinase inhibitor, our lab is focusing on deciphering key molecular targets that are necessary for optimal drug activity. To this end we are expanding the collection of small molecule kinase inhibitors and developing A51-based protelolysis targeting chimera proteins (PROTACs) for degrading critical AML therapeutic target proteins of A51. As patient treatment with many kinase inhibitors often leads to drug resistance, we are already exploring genetic and epigenetic alternations, as well as the immune effectors possibly involved in emerging resistance to A51 treatment. Based on these studies we will design combination therapies for enhancing the efficacy and avoiding resistance to A51. We are also testing the effect of these kinase inhibors in mouse models of Melanoma and CRC.
Evaluating the therapeutic potential of small molecule inhibitors which glue CKIα to ubiquitin ligase cereblon for degradation
Advanced colorectal cancer (CRC) is often an incurable disease. To assess whether the anti-tumorigenic effect of CKIα inhibition can be translated to a new therapeutic approach in CRC, we continue to develop new small molecule CKIα inhibitors compounds and test them in an ex-vivo minigut model of intestinal organoids and in several CRC mouse models. A highly inteseting class of inhibitors is thalidomide derivatives (IMiDs), small molecules which function as a molecular glue, linking the Cereblon ubiquitin ligase complex to CKIα. This is a new promising technology enabling the destruction of protein substrates that by themselves are not recognized and destroyed by the ubiquitin-proteasome pathway. We have already tested the effect of a few CKIα-glues in tumor organoids derived from adenomas of APCmin/- mice (a CRC mouse model mimicking the hereditary CRC syndrome - human familial adenomatous polyposis coli) and observed a marked susceptibility to the inhibitors, with Prox1 downregulation, p53 activation and selective tumor organoid death. Now we move to testing the glue-inhibitors in the APC+/min and the inducible APCΔcolon (APCfl/fl CDX2-CreERT2) CRC mouse models. In addition, we developed a third new mouse model based on colonoscopy-assisted orthotopic implantation of tumor organoids into the colonic submucosa, subject to monitoring tumor growth under treatment by repeated mouse colonoscopy. Preliminary data show that at least one of the new CKIα-glue inhibitor is effective in inducing p53 activation and tumor apoptosis in vivo.
