Our laboratory studies the genetic mechanisms that determine tumor growth and metastasis. In order for tumors to grow and spread throughout the body, cancer cells must acquire various abnormal functions, and this largely occurs through changes in gene activity.
We are interested in the distinctive traits acquired by cancer cells and in the genes that confer these traits.
As a healthy cell undergoes mutations and other progressive changes that lead it to multiply and generate a tumor, built-in mechanisms within cells act to prevent this outcome, providing protection from the disease. Central among these mechanisms is cellular senescence – a coordinated program activated in damaged or premalignant cells, which prevents them from further proliferation, thereby blocking cancer development. It is now recognized that senescent cells that remain viable in tissues can have important effects on a variety of diseases, including on aging. Furthermore, in cancer, the presence of these cells can exert a variety of complex effects on the progression of the disease. Our research aims to understand how cellular senescence influences cell and tissue function as well as on cancer development, and to study how novel drugs that target senescent cells can be utilized.
As a malignant tumor develops, cells within the tumor are not identical to one another: they assume different traits and identities that are driven by a host of processes. Cell heterogeneity and plasticity provide a great advantage to the tumor as it develops, and often determine treatment outcome.
We are studying how malignant breast cancer cells assume different identities and the influence this has on their ability to grow, metastasize, and respond to drug treatment.
A. Cellular senescence and its effects on tissue function and cancer
Upon experiencing damage or cancer-driving mutations, cells often respond by undergoing cellular senescence. In addition to blocking cell proliferation, senescence leads to dramatic changes in cell morphology and metabolism, and often leads to the secretion of a host of factors that can influence neighboring cells and the microenvironment.
It is now understood that senescent cells accumulate with age in multiple organs and appear in the context of many diseases. These senescent cells exert complex cell autonomous and non-autonomous effects that are detrimental to tissue function and their presence can exacerbate disease. Newly identified senolytic drugs (treatments that selectively clear senescent cells) offer the opportunity to target senescent cells for therapeutic benefit.
We are investigating multiple aspects of the physiologic functions of senescent cells.
Our main interests are:
The functions of senescent cells in tumor lesions: At what stages of tumorigenesis are senescent cells formed? In which compartments of the tumor and its stroma are they located? Do senescent cells appear in response to cancer treatments? Once formed, do senescent cells accelerate or suppress tumorigenesis? We are addressing these questions in several cancer types, collecting and characterizing these cells. We are also using our genetically engineered mouse models to induce or eliminate senescent cells from different tumor compartments to test their effects on tumor progression.
One of our main interests is the roles of senescent cells in pancreatic tumors. We found that human pancreatic carcinomas, as well as mouse models of the disease, harbor subset of cells that are in a senescent state. These senescent cells play a substantial role in promoting tumorigenesis. At early disease stages, senescent tumor cells contribute to the generation of an inflammatory state that supports the progression of the disease (Kolodkin-Gal et al, Gut 2022). In advanced tumors, we identified senescent cancer associated fibroblasts (CAFs) present in the stroma, which act to suppress the activity of the immune system against the tumor (Assouline et al, Nature Comm 2024). Specifically, these CAFs repress the activity of cytotoxic CD8+ T cells. We found that treatment with a senolytic drug, ABT-199, removes senescent CAFs and causes CD8+ T cell activation. Importantly, the senolytic treatment potentiates immunotherapy effects in tumor-bearing mice. This suggests that senolytic drugs could be applied to enhance immunotherapy in this disease.

a) Senescent cells detected in pancreatic premalignant PanIN lesions in a Kras-driven mouse model, by staining for senescence associated b-galactosidase (left, blue) or by p16 (right, brown). b) The Cox2 inflammatory protein (green) is expressed in senescent cells in these lesions, which are not labelled by the cell proliferation marker Mcm7 (red). c) Senescent fibroblasts marked by p16 (brown) in mouse (left) and human (right) pancreatic cancers. d) Senescent p16+ cells (red) among CAFs (Pdpn+, green).
The effects of senescent cell accumulation on healthy tissues: How does the prolonged presence of senescent cells in tissues influence their aging rates and propensity for cancer? We developed mice in which molecular activators of senescence – p16Ink4a and p14Arf – can be induced in tissues of choice, allowing us to test the effects of senescence on different organs. Focusing on the epidermis as a model epithelial organ, we discovered that senescent cells, once formed, remain in the tissue for weeks (Tokarsky-Amiel et al., Cancer Res 2013). We subsequently showed that the chronic presence of p16Ink4a-expressing cells in the epidermis paradoxically induces hyperplasia and dysplasia, and promotes epidermal tumorigenesis, through activation of a Wnt pathway feed-forward loop (Azazmeh et al., Nature Comm 2020). This work highlights how senescence can enhance early tumorigenesis through paracrine stimulation of neighboring cells, and the potential of senolytics to prevent progression of the disease. We are exploring the physiologic contexts in which such accumulation enhances tumorigenesis in various organs.
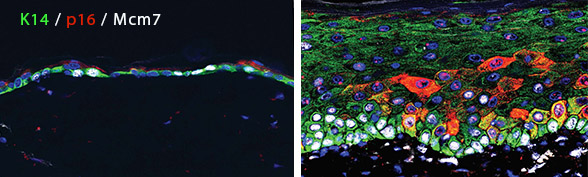
Epidermis harboring cells overexpressing p16 for six months (red, right image) shows layer expansion and hyperplasia, with high levels of cell division (labeled by Mcm7, white). Left image shows normal mouse skin.
Targeting senescent cells: Pharmacologic elimination of senescent cells in conditions where they contribute to disease is a promising new research avenue. Several senolytic drugs that preferentially eliminate senescent cells have been identified in recent years. We established that such drugs are effective in eliminating senescent cells from the epidermis (Yosef et al., Nature Comm 2016). In the skin, senolytic treatment partially reversed epidermal hyperplasia (Azazmeh et al., Nature Comm 2020). In pancreatic cancers, senolytic treatment delayed malignant progression, and activated anti-tumor immune responses (Kolodkin-Gal et al., Gut 2020, Assouline et al, Nature Comm 2024).
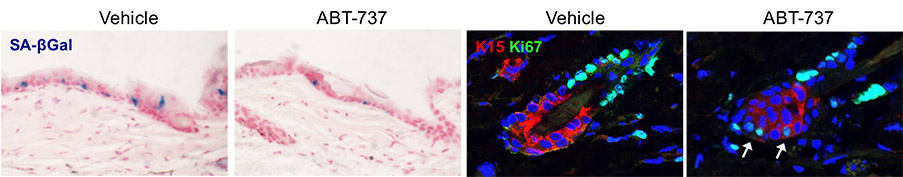
ABT-737, an inhibitor of the Bcl2 protein family, eliminates senescent cells (blue, left images)) induced by p14Arf in the epidermis. b) Increased cell proliferation of hair follicle stem cells upon senolytic treatment. c) Senescent p16+ cells in epidermal hyperplasia in mice, either untreated (left) or treated with ABT-737 (right). d) Senescent p16+ CAFs (red) in implanted mouse pancreatic tumors (YFP), in untreated mice or mice treated with the senolytic drug ABT-199 which targets Bcl2.
Senescence in pancreatic beta-cells: Expression of the senescence activator p16increases with age in pancreatic insulin-secreting beta-cells, yet the consequences of p16 activity in these cells are unclear. We found that p16-induced senescence enhances the ability of beta-cells to execute their major function – secretion of insulin in response to glucose (Helman et al., Nature Med 2016). We analyzed human single-cell mRNA-seq data and identified a subset of p16+ senescent beta cells in adults. These data similarly indicated that these cells acquire a functionally mature state (Patra et al., NAR 2024). This highlights a novel role for senescence and p16 in functional maturation of these cells. We identified another important hallmark of these senescent beta cells: elevated interferon responsiveness. We found that senescent beta cells harbor cytoplasmic DNA foci, and that these generate increased sensitivity to interferon activity, resulting in elevated HLA expression. We are exploring the roles of beta-cell senescence in glucose metabolism with age and in diabetes.
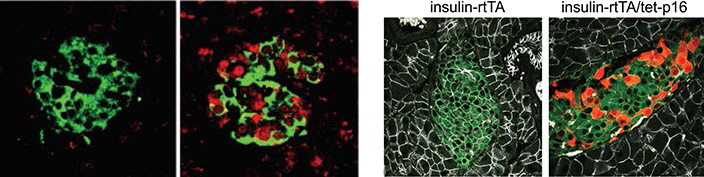
a) Pancreatic islets of a middle-aged human subject, stained for p16 (red), and Insulin (green). b) Transgenic induction of p16 (red) in pancreatic islets of mice (image on left shows control mouse islet). Green labels Insulin. c) UMAP of human pancreatic beta cells, with red indicating expression of p16 (CDKN2A). d) Sections of human pancreatic islets stained for p16 and for HLA-I, indicating increased HLA in p16+ beta cells. e) Cytoplasmic DNA puncta (green, arrows) in senescent beta cells in culture.
B. Cellular heterogeneity in breast cancer: regulation of differentiation state transitions
Human tumors are highly heterogeneous, differing widely in their characteristics and their severity. However, even within a single tumor, individual cells typically show a range of identities and traits. This intratumoral cellular heterogeneity supports the ability of the tumor to grow and disseminate, and, most importantly, underlies the ability of cell subsets to avoid death upon therapy. One important mechanism by which cancer cells change identity is by modifying their differentiation state, often through the activity of central regulatory pathways. Our lab is interested in mechanisms controlling the differentiation state of breast cancer cells: Which molecular drivers dictate different states, including stem- and progenitor-like states? How do cells switch identities? How is tumor cell differentiation state responsive to external signals? How does differentiation state affect the ability of tumor cells to grow, disseminate, and resist therapy?
Our work has uncovered roles for specific regulators such as TCF3, EZH2, FOXA1, NOTCH and others in regulating cell state in triple-negative breast cancer, and revealed the occurrence of asymmetric divisions generating cellular heterogeneity (Granit et al Cell Rep 2018, Granit et al., Oncogene 2013, Slyper et al Cancer Res 2012).
Left: a human triple-negative breast cancer showing heterogenic cellular expression of K18 (green) and K14 (red). Middle: a triple-negative monoclonal tumorsphere showing heterogenous expression of the same markers, demonstrating plasticity. Right: an asymmetric division demonstrating differential expression of the same markers in daughter cells.
