Our Research
We study how genetic and epigenetic alterations of regulatory DNA elements cause cancer or contribute to the disease. We focus on two types of regulatory DNA elements: enhancers (regulating transcription), and CTCF binding sites (regulating chromosomal topology, i.e. the folding of the chromosome in 3D).
Epigenetic topological alterations
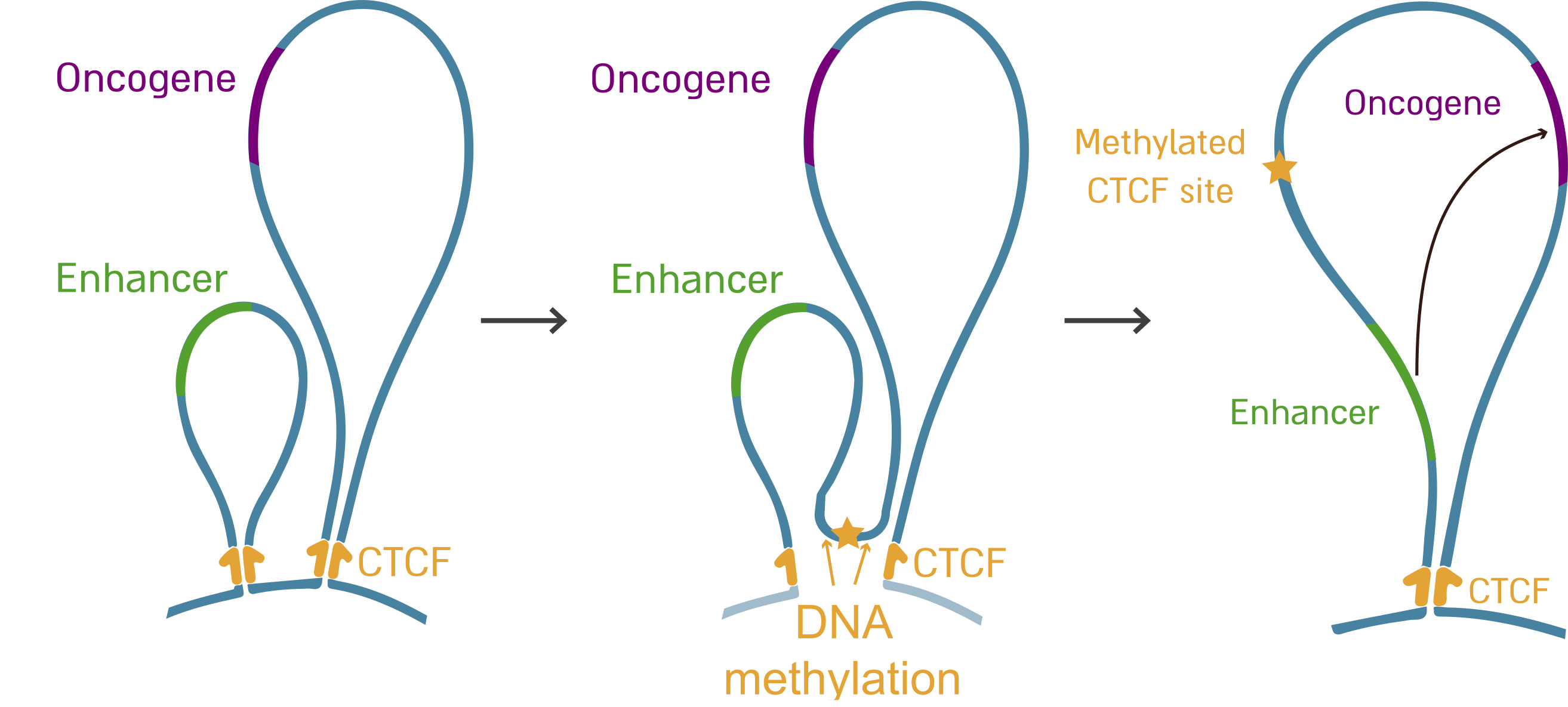
Normally, our chromosomes are divided into multiple topological domains that allow frequent interactions within each domain, but limit the interactions across domain boundaries. We have previously demonstrated that aberrant DNA methylation of CTCF binding sites perturbs chromosomal topology in IDH mutant glioma (Flavahan*, Drier* et al. Nature 2016) and SDH deficient gastrointestinal stromal tumors (Flavahan*, Drier* et al. Nature 2019). We demonstrated that in these tumors, CTCF binding sites at boundaries get methylated, lose CTCF binding, disrupt insulation between adjacent topological domains, and allow aberrant interactions between genes and enhancers. Specifically, this allows activation of the PDGFRA oncogene in gliomas and FGF3, FGF4 and KIT in gastrointestinal stromal tumors, and therefore promotes tumorigenesis. This groundbreaking model demonstrates that epigenetic and topologic alterations can drive cancer, while highlighting the importance of regulatory DNA alterations.
We are extending this framework to additional types of cancer and of topologic disruptions to test the interplay between metabolism, epigenetics, topology and gene regulation, and how it is dysregulated across different types of cancer.
Transcriptional, epigenetic and topologic intratumor heterogeneity

We develop systematic approaches to infer and integrate genetic, epigenetic, regulatory, transcriptional topologic and functional information in the single cell level, to characterize and study the relationships between different types of subclones within a given tumor. We developed a method to predict pathway activity in single cells from scRNA-seq to support these efforts (SiPSiC, Davis, Wizel and Drier, Genome Research 2024). We are studying how transcriptional and regulatory heterogeneity drive disease and response to therapy, for example in adenoid cystic carcinoma (Parikh*, Wizel* et al. Cell Reports 2022), HPV-related multiphenotypic sinonasal carcinoma (Wizel*, Spence* et al. 2025), and acute lymphoblastic leukemia (Anand*, Guillaumet-Adkins*, Dimitrova*, Yun*, Drier* et al. Blood 2021).
The role of SMCHD1 and CTCFL in regulating chromosome interactions, gene expression and splicing
SMCHD1 is a non-canonical SMC
protein and an epigenetic regulator. Loss of SMCHD1 perturbs histone
modifications, DNA methylation, CTCF binding and chromosomal interactions, but
the mechanism of action is not clear. Mutations in SMCHD1 lead to two distinct
genetic diseases- facioscapulohumeral muscular dystrophy (FSHD) and
Bosma-arrhinia and microphthalmia syndrome (BAMS). CTCFL is a paralog of
CTCF, sharing a very similar DNA binding domain, but divergent N- and
C-terminal domains. Normally CTCFL is expressed only in germ cells, however it
is also aberrantly expressed in several cancers. CTCFL is also believed to
disrupt CTCF binding at the sites it binds to. We study how mutations and
aberrant levels of SMCHD1 and CTCFL impact CTCF binding, chromosomal interactions,
gene expression and splicing and how all this contributes to development of
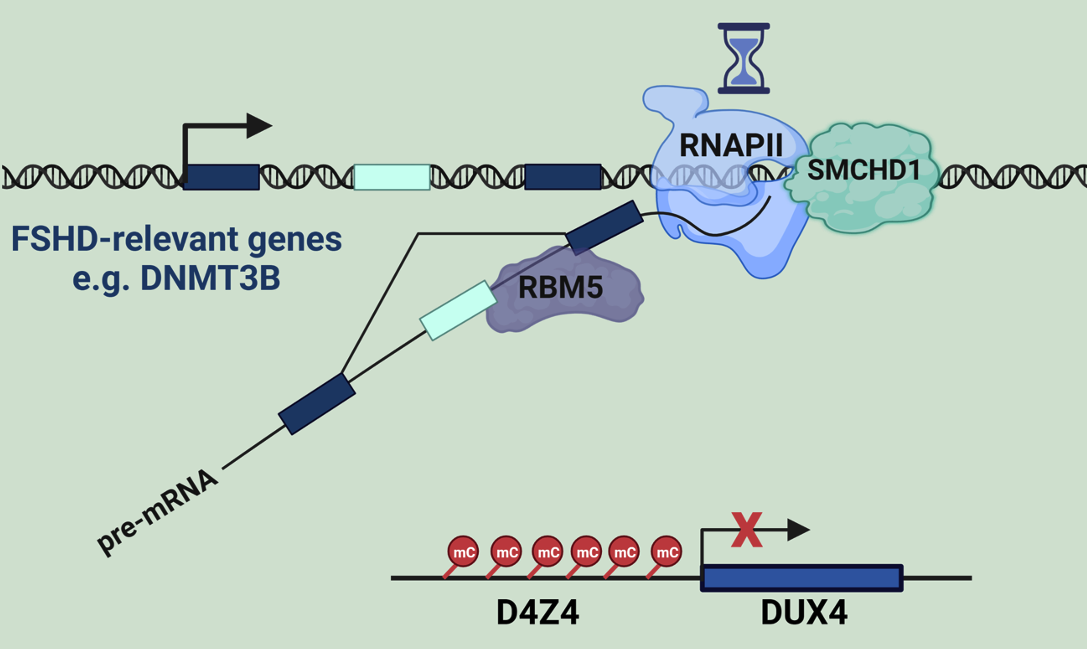
cancer, FSHD and BAMS. For example, we recently demonstrated that loss of
SMCHD1 leads to mis-splicing of DNMT3B, lower methylation at the D4Z4
macrosatellite repeat, and overexpression of DUX4, the factor that drives
muscle cell death in FSHD (Engal et al. Science Advances 2024).
Click here to see selected publications.
